A Fibrose Quística é uma doença rara, genética e degenerativa, que afeta cerca de 80.000 pessoas em todo o mundo e cerca de 400 em Portugal A Dra. Pilar Azevedo, pneumologista do Centro de Referência em FQ de Santa Maria e coordenadora do recém-criado Grupo Nacional de Estudos de Fibrose Quística (FQ) – um espaço de discussão sobre possíveis áreas de investigação, debate de experiências e aposta numa rede nacional que visa melhorar o tratamento de pacientes – explica-nos o que é esta doença, as dificuldades com que se debatem os doentes e as terapêuticas existentes atualmente no intuito de melhor e prolongar a sua qualidade de vida.
A raridade, complexidade e especificidade do tratamento desta doença exige a criação de equipas multidisciplinares diferenciadas, que contemplem desde médicos de Pediatria e Pneumologia, enfermeiros, nutricionistas, psicólogos e outros técnicos de saúde fundamentais na avaliação, apoio e acompanhamento do paciente com FQ.
O que é a Fibrose Quística?
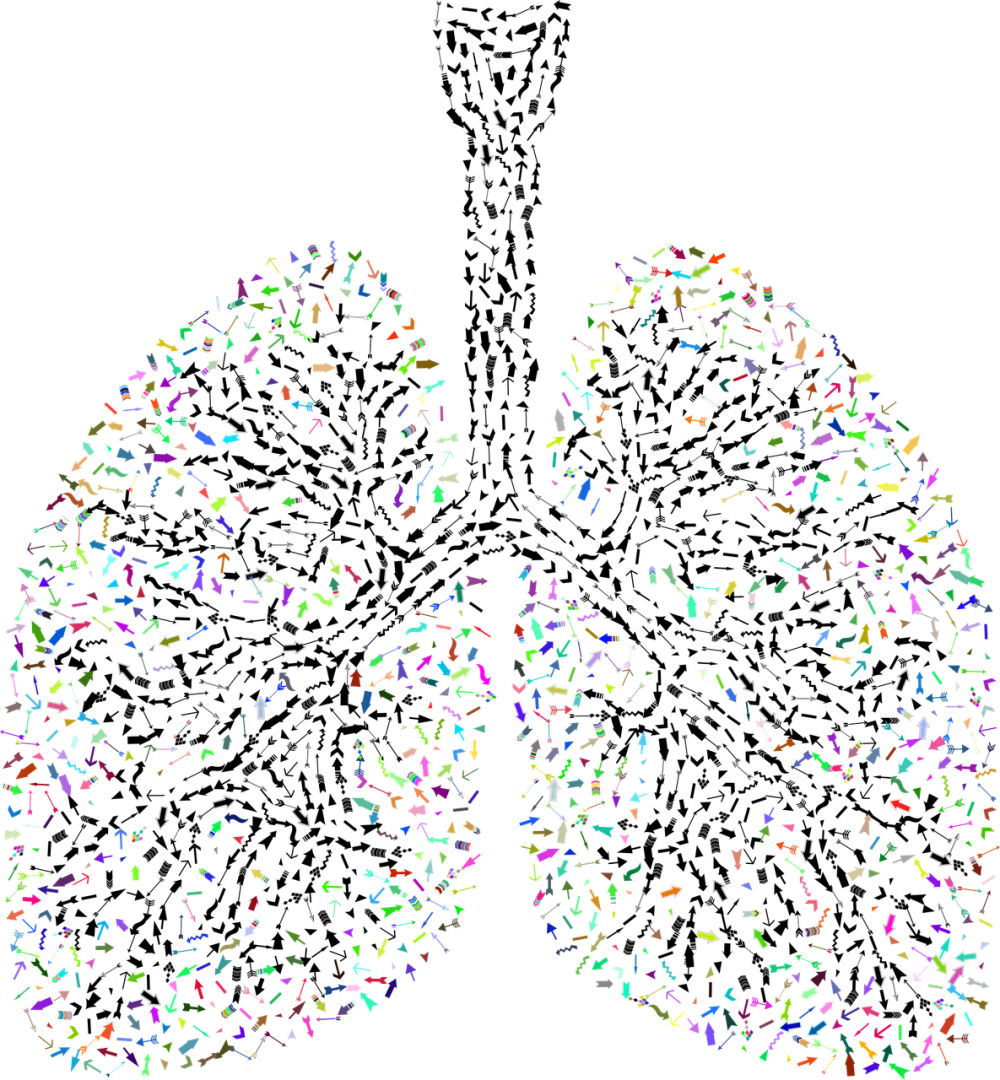
A Fibrose Quística é uma doença genética, hereditária, que passa de pais para filhos. O que acontece nesta doença é a alteração de um gene, o gene CFTR, responsável pela codificação da proteína CFTR. Esta proteína está nas células de vários órgãos, sendo que a doença se expressa mais, nos pulmões, no pâncreas, no fígado, no intestino, no aparelho reprodutor e na pele. É pois uma doença que envolve várias partes do corpo, mas que afeta sobretudo estes órgãos.
A Fibrose Quística é uma doença que está descrita desde 1930. Nessa altura os doentes morriam precocemente. Foram sendo descobertas novas terapêuticas e os doentes passaram a ser tratados em centros especializados o que melhorou o prognóstico e aumentou a esperança de vida.
Em 1989, descobriram que havia um gene responsável pela doença, o que foi uma descoberta extraordinária até porque se pensava que se poderia vir a descobrir um tratamento genético. Mas, entretanto, percebeu-se que há imensas mutações estando atualmente descritas cerca de 2107. Destas só um número muito reduzido é que tem uma expressão clínica identificada (cerca de 20 a 25). Mesmo dentro deste número reduzido, a maioria são muito raras, mas existe uma mutação, a F508del, que é muito frequente sendo que cerca de 90% dos doentes tem esta mutação. Para se ter esta doença a pessoa tem de ter duas mutações. De todas as mutações, algumas causam formas muito graves de doença e outras causam formas mais ligeiras.
Quando dizemos que alguém tem Fibrose Quística não quer dizer que tenha sempre a mesma expressão clínica, porque depende das mutações, da influência de genes modificadores e de fatores do ambiente tais como o tipo de vida que a pessoa leva e as condições em que vive. Portanto, o diagnóstico desta doença não significa que a pessoa tenha sempre o mesmo tipo de quadro clínico.
Como é que é adquirida? Geneticamente?
É uma doença genética e apesar de ser rara, é uma das mais comuns e frequentes no Norte da Europa. É uma doença autossómica recessiva, em que que ambos os pais têm que ser portadores ou doentes para terem filhos com a doença. Mas é importante referir que os pais não tem necessariamente de ter a doença, podem apenas ser portadores da anomalia genética.
Como podemos saber se somos portadores da anomalia genética da Fibrose Quística? Não devia este ser um teste a ter em conta em quem pretende ter filhos?
Geralmente não se procuram portadores. Essa procura só se faz quando existe um caso de Fibrose Quística na família.
Em Portugal, desde 2013, temos o rastreio neonatal da Fibrose Quística, que começou com um projeto piloto através do teste do pezinho. Os resultados foram bons e, em 2018, introduziu-se o rastreio da Fibrose Quística, no Rastreio Nacional Neonatal. Todas as crianças que nascem em Portugal são testadas. Mas isto só a partir de 2013, portanto temos algumas pessoas que, nascendo anteriormente, podem não estar diagnosticadas. As pessoas que ainda não estão diagnosticadas, principalmente aquelas que têm formas atípicas de manifestação da doença, acabam por ser uma preocupação para nós, porque queremos diagnosticar todos os doentes.
Com os dados que temos desde 2013, estimamos que a nossa prevalência seja de 1 para 8 mil recém-nascidos. Se compararmos os nossos dados com os de Espanha, temos menor prevalência de Fibrose Quística do que os espanhóis, o que não deve ser verdade. Por isso ainda temos de analisar melhor os nossos dados, para termos uma noção mais clara de qual é a real prevalência da Fibrose Quística em Portugal.
Em relação à testagem antes de se ter filhos, não se justifica se não existir um historial de Fibrose Quística na família, mas as pessoas com história familiar de Fibrose Quística devem ser testadas para saber se são portadoras.
Trata-se de uma doença que é rara e em Portugal estão descritos cerca de 400 doentes numa população de aproximadamente 10 milhões de habitantes. No mundo inteiro existem cerca de 75 000 doentes.
– Quantos doentes existem em Portugal e qual a média estimada de portadores do gene? Como é feito o diagnóstico atualmente?
Em Portugal estão descritos cerca de 400 doentes como já disse.
O diagnóstico é feito através do rastreio neonatal para quem nasce depois de 2013 sendo os casos positivos encaminhados para exames de confirmação. Para quem nasceu antes deste ano e no caso de haver suspeita clínica, são também realizados exames de confirmação de diagnóstico como a prova e suor e o estudo genético. É importante que os médicos estejam sensibilizados para a existência desta doença.
Quando existe, por exemplo, no mesmo doente uma associação de problemas respiratórios e gastrointestinais ou em alguns casos de infertilidade no homem adulto, podemos suspeitar da existência da doença e pedir um primeiro teste de diagnóstico que é a prova do suor. Estes doentes têm tipicamente um suor muito salgado, devido às alterações da proteína CFTR na pele, que faz com que se acumule cloreto de sódio no suor. Se esta prova for positiva, avança-se para o estudo genético, de forma a encontrar as mutações que permitem fazer o diagnóstico.
Quando a dúvida prevalece existem métodos mais sofisticados e ainda numa fase de validação, para estudar a função da proteína no epitélio nasal ou na mucosa retal. São métodos de diagnóstico sofisticados, realizados em laboratórios trabalhando em colaboração com os centros de referência.
No dia-a-dia, quais as limitações comuns a estes doentes?
Vamos supor um cenário de um caso mais grave. Uma criança muito pequenina que começa logo com problemas respiratórios, com secreções brônquicas muito espessas e difíceis de mobilizar que parecem lama. Nestas secreções espessas e retidas nas vias aéreas começam a crescer bactérias, e o doente começa a estar sempre com infeções respiratórias e com muita tosse e muita expetoração. Como consequência os doentes acabam por estar sempre internados ou terão que fazer muitas vezes antibióticos em casa. Quando o pâncreas também está afetado, os doentes não fazem a digestão adequadamente, e têm diarreias muito graves em que perdem muitos nutrientes conduzindo a atrasos no crescimento, emagrecimento muito acentuado, deficiência de vitaminas lipossolúveis. Por vezes também surgem problemas nos intestinos muito graves.
Ou seja, são pessoas que vão ter sempre limitações, porque estão quase sempre doentes sobretudo com infeções respiratórias e problemas pulmonares graves. As crianças faltam muito à escola, não brincam como as outras crianças, têm dificuldade em socializar porque é preciso cuidado para que não apanhem infeções, o atraso no crescimento dificulta a integração na vida normal e tudo isto vai-se agravando à medida que o doente cresce. A doença pode progredir de forma tão grave que a terapêutica médica pode não ser suficiente e temos de referenciar o doente para transplante pulmonar.
Em Portugal temos um excelente centro de transplante pulmonar, no hospital de Santa Marta, com uma taxa de eficiência equiparável a qualquer centro europeu. Foi a nossa grande ajuda até agora. Temos um número significativo de pessoas com Fibrose Quística transplantadas e a sobrevida do transplante pulmonar na Fibrose Quística é bastante boa e até melhor do que noutras doenças. A qualidade de vida melhora significativamente, após o transplante embora uma pessoa transplantada acabe também por ter outras limitações resultantes da terapêutica imunossupressora.
Como é feito o acompanhamento dos doentes em Portugal? E que lacunas ainda existem a este nível?
Em Portugal temos centros de referência, o que é ótimo.
Nos Estados Unidos, entre os anos 90 e 95, percebeu-se que os doentes que eram seguidos em centros especializados tinham uma evolução muito melhor e por isso foi defendido que as pessoas com Fibrose Quística deviam ser seguidas por especialistas que conhecessem bem esta doença.
Os europeus viram o exemplo norte-americano e reconheceram que os resultados eram efetivamente muito melhores e defenderam também a criação de centros de referência na Europa, os quais concentravam no mesmo espaço tudo o que fosse necessário para o diagnóstico e tratamento dos doentes por profissionais especializados.
Em Portugal temos cinco centros de referência reconhecidos pela DGS, dos quais dois em Lisboa, dois no Porto e um em Coimbra. Estes centros têm sempre um polo pediátrico e um polo de adultos.
Estes centros de referência têm um modelo muito interessante. A criança é diagnosticada e é seguida no centro pediátrico até aos 18 anos.
O centro pediátrico garante uma abordagem multidisciplinar com médicos pediatras vocacionados para tratar estas crianças, enfermeiros, entre eles enfermeiros especialistas em reabilitação e também fisioterapeutas porque estas crianças têm de fazer muita reabilitação respiratória. As equipas incluem ainda psicólogos, porque é uma doença com muito impacto quer na criança, quer na família, assistentes sociais para apoiar as famílias esclarecendo-as sobre os benefícios sociais a que têm direito, nutricionistas e dietistas. Portanto são equipas com muitos profissionais sendo o doente seguido por todas estas pessoas, que se reúnem regularmente para discutirem os casos.
Quando o doente atinge os 18 anos inicia-se a fase de transição dos cuidados pediátricos para os cuidados de adultos. É uma fase em que, se não surgirem problemas graves, os doentes entram na universidade ou no mercado de trabalho, querem viajar, fazer Erasmus, começam a trabalhar, a namorar, a ter vontade de constituir família… Nesta altura é quando se inicia a transferência para o centro de adultos. Para que esta transferência não seja abrupta, existem programas de transição muito bem definidos. Os pediatras que seguiam estes doentes e os médicos de adultos, geralmente Pneumologistas, que passarão a fazer o acompanhamento, vão trabalhar em conjunto durante algum tempo até sentirem que o doente e a família estão preparadas para a transição para o centro de adultos.
No centro de adultos temos exatamente a mesma organização. Como já disse os médicos geralmente são pneumologistas, porque o órgão mais afetado é o pulmão e a insuficiência respiratória é a principal causa de morte nesta doença. O resto da equipa tem a mesma constituição da do centro pediátrico sendo os procedimentos e os protocolos também idênticos. Há consultores de várias especialidades que colaboram com o centro como gastroenterologistas, diabetologistas, otorrinolaringologistas, ginecologistas.
Estes centros incluem os dados dos doentes num registo europeu de Fibrose Quística, o que permite haver mais informação sobre a doença, uma vez que quantos mais doentes analisarmos, mais conseguimos perceber a doença. Com estes dados é também possível olhar para os resultados no final de cada ano e ver como é que Portugal está em comparação com os outros países europeus e inferir se estamos a trabalhar no caminho certo.
O centro de referência de Fibrose Quística de Santa Maria pertence à rede europeia de ensaios clínicos e os grandes ensaios clínicos relacionados com a Fibrose Quística que são propostos pela Sociedade Europeia de Fibrose Quística também passam por nós. Portugal tem a grande vantagem de ter uma investigação básica nesta área de grande qualidade. Temos investigadores na área da Ciência Básica em Fibrose Quística reconhecidos internacionalmente e com grande prestígio científico.
O Covid-19 veio implicar ainda maior limitações na vida destas pessoas?
Como todas as patologias, na fase inicial da pandemia, sim. Não se interrompeu o acompanhamento dos doentes mas houve algumas dificuldades. A telemedicina, com as consultas online e por telefone ajudaram. Acabaram por se desenvolver um conjunto de estratégias no sentido de manter os doentes sempre vigiados.
Um doente com Fibrose Quística, por norma, é visto de 2 em 2 meses. No dia em que vai ao centro é visto pelo médico e todos os outros profissionais de saúde, faz todas as análises, o exame da expetoração e faz avaliação da função respiratória.
Com a pandemia, durante um curto período de tempo, os doentes não iam ao centro e nós fazíamos tudo de forma não presencial. Mas rapidamente todos os centros criaram as condições de segurança necessárias e os doentes voltaram a ser novamente seguidos.
Também temos uma estratégia de hospitalização domiciliária. Quando os doentes precisam de fazer antibiótico endovenoso, que é muito frequente, fazem-no habitualmente no regime de hospital de dia. Assinam o consentimento informado, nós ensinamos a fazer a antibioterapia e assim conseguimos que o doente faça a terapia em casa. Esta alternativa é uma vantagem em relação aos internamentos, que por norma duram 2 semanas, diminuindo os custos e os riscos associados ao internamento e contribuindo para melhorar a qualidade de vida dos doentes.
No caso das crianças, estão as escolas preparadas para dar respostas às suas necessidades?
Não poria o problema nesses termos. A escola é um espaço para a criança aprender, socializar e ser estimulada.
As escolas têm poucas coisas a fazer para se prepararem mais do que já estão para uma doença destas. Não são em regra necessárias grandes intervenções quando surge uma criança com Fibrose Quística.
Obviamente que os pais têm de falar com os professores e com todos os profissionais da escola, porque são crianças que precisam de cuidados específicos, como uma alimentação mais cuidada, tomar regularmente compridos que facilitam a digestão, ou fazer insulina entre outros condicionantes.
São crianças que tossem muito, podem ter cansaço e falta de ar e não aguentar as aulas de educação física ou as brincadeiras no recreio, mas nada que uma escola em que os professores e auxiliares que estejam informados e atentos não possam ajudar a resolver. Sempre que há uma criança doente existem mais exigências, mas há limitações como a surdez, a cegueira, em que são precisas adaptações muito maiores.
Como é a evolução da doença? As doenças do foro pneumológico são as responsáveis pela morte precoce devido a esta doença?
Sim, a principal causa de morte é pulmonar. A morte geralmente ocorre como consequência da lesão pulmonar que pode ser gravíssima.
Como era feito o tratamento até agora e o que muda com a introdução do Kaftrio em Portugal?
Antes do Kaftrio limitávamo-nos a tratar os sintomas.
Sendo os pulmões os órgãos mais afetados, os doentes têm muitas infeções respiratórias e as secreções são muito espessas e viscosas. Por isso estes doentes têm de fazer uma terapêutica inalada com fármacos que fluidificam as secreções e depois fazem a ginástica respiratória para expelir essas secreções. Alguns doentes fazem também antibióticos por via inalatória.
É uma terapêutica exaustiva e que ocupa grande parte do dia. E isto só considerando os sintomas respiratórios. Quem tem problemas gastrointestinais e do pâncreas tem de tomar comprimidos e vitaminas. Quem tem diabetes relacionada com a Fibrose Quística tem de fazer insulina. É uma carga terapêutica muito pesada.
Em 1989 quando descobriram o gene achava-se que se podia tratar a doença com terapêutica genética. No entanto, é muito difícil chegar ao gene que está no núcleo da célula, pelo que se percebeu que a terapêutica genética seria difícil de implementar.
Mas este gene produz uma proteína que funciona mal, e como é difícil chegar ao gene e corrigi-lo, podemos mais facilmente chegar à proteína e corrigir o seu defeito. Desenvolveram-se, assim, moléculas para corrigir a proteína, o que veio modificar o paradigma de tratamento desta doença e pode mesmo servir de exemplo para outras doenças. Nós não estamos agora a tratar sintomas, mas estamos a corrigir o defeito que provoca esses sintomas ou seja, pomos a proteína a funcionar bem.
Em 2012 surge a primeira molécula, cujo nome comercial era Kalydeco, que só era dirigida a umas mutações raríssimas que correspondiam a apenas 1 % dos doentes com Fibrose Quística. Mas teve muito sucesso. No entanto, tratávamos apenas a um grupo restrito de doentes. Precisávamos de tratar os doentes com a mutação mais frequente que é a F508del que são a maioria.
Em 2015 surge então outro fármaco com duas moléculas, que já era direcionado a quem tinha duas mutações F508del. No entanto os resultados eram muito piores do que aqueles conseguidos com a primeira molécula.
Em 2018 modificam o fármaco que passou a conseguir atingir também os doentes que tivessem apenas uma mutação F508del, para além dos anteriores, ou seja, alargámos o número de doentes que poderiam beneficiar desta terapêutica. Mas os resultados continuavam a ser semelhantes aos anteriores.
E agora surgiu a terapêutica tripla dirigida a todos os doentes que têm pelo menos uma mutação F508del com a qual atingimos quase 90% dos doentes com Fibrose Quística. Esta terapêutica teve resultados incríveis. Os doentes recuperam a função pulmonar de uma forma significativa. Todos os doentes referem que deixam praticamente de ter tosse ou expetoração, que têm mais energia e que conseguem fazer uma vida normal ou quase normal. É surpreendente!
Quando fazemos as provas funcionais para vermos os resultados de uma forma mais objetiva, comprovamos que efetivamente melhoram de uma maneira incrível. Quando fazemos as TACs também se comprova a melhoria. Os doentes ganham muito peso e começam a ter uma vida normal e a não necessitar praticamente de manter as terapêuticas sintomáticas que faziam. Conseguimos transformar a Fibrose Quística, numa doença crónica, que conseguimos tratar com alguns comprimidos.
Se a estes avanços terapêuticos juntarmos a possibilidade que temos agora de fazer o diagnóstico à nascença recorrendo ao rastreio neonatal, é fácil perceber que ocorreram mudanças significativas na abordagem da Fibrose Quística, porque podemos começar um tratamento muito eficaz de correção do defeito antes de existirem alterações irreversíveis. Hoje em dia sabemos que o doente com Fibrose Quística pode viver muitos anos e viremos a ter idosos com esta doença.
Mas como a doença não depende apenas do genótipo CFTR mas depende também de genes modificadores e de fatores ambientais, há casos em que o doente pode responder muito bem às novas terapêuticas e outros em que não responderá tão bem e pode inclusivamente ter efeitos acessórios.
Por isso há todo um trabalho importante a ser feito com a Ciência Básica para se identificar os doentes potencialmente respondedores.
Presentemente estamos a fazer estudos de vida real com estes novos fármacos para os conhecermos melhor e analisarmos a sua eficácia e segurança ao longo do tempo.
Já foram feitos os estudos necessários pela indústria farmacêutica e pelas entidades reguladoras para avaliar a a eficácia e segurança dos fármacos e agora nós médicos avaliamos os resultados na vida real. E com o Kaftrio, que já estamos a utilizar há 1 ano em Portugal, os resultados têm sido muito bons.
Ainda não temos tratamento para os doentes que não têm as mutações mais frequentes, mas a investigação continua e estão a ser estudadas outras terapêuticas para estes doentes. Inclusivamente os fármacos já existentes estão a ser melhorados.
Portanto se há doença que tem no seu horizonte uma esperança enorme é a Fibrose Quística e neste momento em Portugal podemos ter acesso a todas estas terapêuticas.